Diseño de antivirales usando computadores cuánticos?
Anti virals drug design using quantic supercomputation?.
By Juan Carlos López Corbalán, MD, Ph.D.,R.Ph. (1); Guillermo García Alcayne, Ph.D. (2)
(1) Subdelegación del Gobierno en Alicante Servicio de Inspección Farmacéutica y Control de Drogas.Alicante España.
(2) Prof.Emérito.Universidad Autónoma Dep.Mécánica Cuánticas.Madrid.
.
INTRODUCTION
Currently the SARS-CoV2 beta coronavirus pandemic has caused more than 6.5 million deaths and 520 million infected, ( Johns Hopkins Coronavirus Resource Center) and the figures will be obsolete in a few minutes, because between 29,000 and 56,000 people are dying every day on the planet. (20)
We believe that supercomputing drug design could be a useful tool to find that cheap, effective, orally administered antiviral with no adverse reactions.
Supercomputing versus quantum computers.
The key point is the unión os SARS CoV2 to the main Spike protein ( 30, 33, 35, 36, 75), with two previous cleavajes.
In silico models are used to search for antiviral drugs. Normally on drugs that have already been evaluated and repositioned, and have passed the tests of Toxicology, Mutagenicity (Ames test, inhibition of ovarian cells in hamsters, for example) and have already been approved by (52,54, 55).
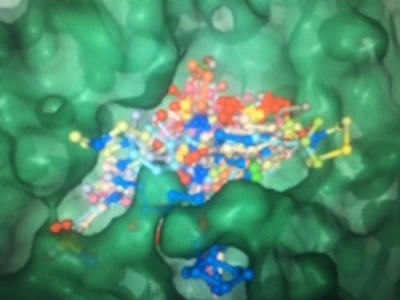 Fig. 1 Hydrofobic residue are ideal áreas for drug binding to the Surface.
The drugs regulatory agencies, although for other indications.
There are pharmacological groups like the ones you can see in the attached figure and by means of computation we can screen theoretical models, looking for hydrophobic residues,
Where would "fit" low molecular weight molecules (drugs with Pm between 350 and 900), such as those marked in and represented in the figure below.
such as those marked in and represented in yellow in this other three-dimensional view.
.Today Nanome Inc has developed VR programs to stydy carefully the hidrofobic residues and binding and then we would measure the entropy, enthalpy and binding energies (expressed in -Kcal/mol).
The more negative the energy, the more electrostable is the binding to the receptor.
Fig. 1 Hydrofobic residue are ideal áreas for drug binding to the Surface.
The drugs regulatory agencies, although for other indications.
There are pharmacological groups like the ones you can see in the attached figure and by means of computation we can screen theoretical models, looking for hydrophobic residues,
Where would "fit" low molecular weight molecules (drugs with Pm between 350 and 900), such as those marked in and represented in the figure below.
such as those marked in and represented in yellow in this other three-dimensional view.
.Today Nanome Inc has developed VR programs to stydy carefully the hidrofobic residues and binding and then we would measure the entropy, enthalpy and binding energies (expressed in -Kcal/mol).
The more negative the energy, the more electrostable is the binding to the receptor.
 . Fig. 2 VR and designo d new drugs ( Nanome Inc.)
This classical process is the Doking". Classical cryptography is dead. Only its new variant, the quantum one, can be considered safe, because the screening of thousands of molecules needs powerful computers and not the usual ones. This is the only way we could test 100,000 compunds a year.
. Fig. 2 VR and designo d new drugs ( Nanome Inc.)
This classical process is the Doking". Classical cryptography is dead. Only its new variant, the quantum one, can be considered safe, because the screening of thousands of molecules needs powerful computers and not the usual ones. This is the only way we could test 100,000 compunds a year.
 .
Fig 3. Electrophysical forces
At the momento there is no effective antiviral treatment against this plague, except for three drugs very recently approved by the FDA:
(a) Molnupiravir (MK4482), a product of Merck & Ridgeback Therapeutics, which is an RNA analogue that is actually a pro-drug since it is transformed in the body into N-hydroxycytidine (NHC).
Indicated for patients with good oxygen saturation, with one or more risk factors. at a dose of 800 mg / 12 hours / 5 days, with an initial RR of 48% which later, as is often the case, was reduced to an RR of 30%.
This initial optimism is very characteristic of current pharmaceutical companies, with a tendency to inflate the data.
Molnupiravir is mutagenic: proven by the Ames test for Salmonella tiphymurium, the HPRT assay in hamster ovary cells and for developing teratogenicity in rats at high doses.
It was approved by the FDA by a narrow vote of 13 to 10.
b) Pfizer's Ritonavir + PF-07321332 combination. 100 mg of Ritonavir (which actually acts as an enzyme motivator9 + 300 mg of PF07321332 or Nirmatrelvir.
It is an Mpro (protease) inhibitor of SARS-CoV2. With a RR of 89% after 5 days of treatment and 85% after 5 days of treatment.
c) Monoclonal antibodies. That is, antibodies generated against a specific epitope of the virus surface. Epitope and antigenic determinant are the same, warning for computer scientists who are designing drugs. Leave the field free to virologists such as Prof. Luis Enjuanes, Mariano Esteban or Vicente Larraga, Isabel Sola, Sofía Zúñiga or Juan García Arriaza.
At the momento of review of this article mosto f Monoclonal antibodies are less active, only Barnmalizumab are useful right now.
Product Absolute risk NNT Teratogenicity
Molnupiravir (30%) 6,7 % 34 Yes6,7 %
FP07321332 (89%) 6,7 % 18 No
Monoclonal Ab (70-80%) 8% 11-25 No
Analyzing with biostatistical parameters, these are the differences.
Facts: Computers do not design ANYTHING if they are not well programmed to do so, but they are very useful and can be used for many purposes:
(a) Computational hit counting.
b) Detecting the three-dimensional arrangement of the minimal features in the form of an electron cloud mapping.
c) Resolution of searching for hydrophobic residues in molecules with Pm below 1500 (ideally between 350 and 900) - d) Interactions with the receptor.
d) Receptor interactions by calculating the binding energies (expressed in Kcal/mol) between these putative receptors for new (or repositioned) drugs, as shown in the figure.
e) Measurement of H (kcal/mol), S (cal/mol Kelvin) and Gibbs Energy G increments.
Short chapter of QM ( MD´s skip this one)
To give you an idea of the difficulties of quantum algorithms let us look at a couple of much simpler examples.
2.1 Shor's algorithm (1984) This algorithm is used to factor large integers.
Shor's theorem is from 1994, for factoring composite numbers.
Shor's algorithm consists of two parts:
Classical part.
We have a pseudo-random number a < N.
Compute the mcd(a, N). This can be done using Euclid's algorithm.
If the mcd(a, N) ≠ 1, then it is a nontrivial factor of N, so we are done.
Otherwise, we use the period-finding subprogram (see below) to find r, the period of the following function:
{displaystyle f(x)=a^{x}}f(x)=a^{x}}{mbox{mod}},
i.e. the smallest integer r for which
{displaystyle f(x+r)=f(x)}f(x+r)=f(x).
If r is odd, we go back to step 1.
If ar/2 ≡ -1 (mod N), we go back to step 1.
The factors of N are the mcd(ar/2 ± 1, N).
Quantum part: subprogram to find the period.
We start with a pair of input and output qubit registers with log2N qubits each, and initialize them to.
{ {{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xright\rangle }{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xright\rangle}\rangle|0right\rangle} }
Construct f(x) as a quantum function and apply it to the above state, to obtain.
{ {{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xrightrangle|f(x)\{rightrangle }{{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xrightrangle|f(x)\{0}}.
We apply the quantum Fourier transform to the input register. The quantum Fourier transform at N points is defined as:
{{displaystyle U_{QFT}\left|x\rightrangle =N^{-1/2}sum _{y}e^{2\pi ixy/N}\left|y\rightrangle }U_{QFT}\left|x\rightrangle =N^{-1/2}sum _{y}e^{2\pi ixy/N}\left|y\rightrangle}.
Which leaves us in the following state:
{displaystyle N^{-1}sum _{x}sum _{y}e^{2\pi ixy/N}\left|yrightrangle|f(x)\N^{-1}sum _{x}sum _{y}e^{2\pi ixy/N}\left|yrightrangle|f(x)\N^{-1}sum _{x}sum _{y}e^{2\pi ixy/N}\left|yrightrangle|f(x)\N^{-1}rangle}.
We obtain a certain result y in the input register and f(x0) in the output register. This step is not necessary since, according to the deferred measurement principle, the result will be the same at the end of the algorithm regardless of whether a measurement is performed. However, for reasons of simplification in understanding the algorithm, we will include this step. Since f is periodic, the probability of measuring a certain y is given by
{{x:\,f(x)=f(x_{0})}e^{2\pi ixy/N}right|^{2}=N^{-1}\left|sum _{b}e^{2\pi i(x_{0}+rb)y/N}right|^{2}}N^{-1}\left|sum _{{x: \,f(x)=f(x_{0})}}e^{{2\pi ixy/N}}\right|^{2}=N^{{-1}}\left|\sum _{{b}}e^{{2\pi i(x_{0}+rb)y/N}}\right|^{2}
The analysis now shows that the higher this probability is, the more yr/N is close to an integer.
We convert y/N to an irreducible fraction, and extract the denominator r ', which is a candidate for r.
We check whether f(x) = f(x + r '). If so we are done.
If not, we obtain more candidates to r using values close to y, or multiples of r '. If any candidate satisfies the conditions, we are done.
If not, we go back to step 1 of the subprogram.
ummarized In the figure above. Factorization of n composite numbers.
Grover's algorithm is used to find a datum in an unordered list:
In other words, it tells us the time we would need for an exhaustive key test, it comes from Quantum mechanics It was invented by Lov K. Grover in 1996.
If an unordered sequence with N components is considered. The algorithm requires an N-dimensional state space H, which can be modeled with log2N qubits.
Let us number the entries of the sequence with 0, 1,.... (N-1); and let us select an observable Ω, acting on H, with N known distinct eigenvalues. Each of the eigenvalues of Ω encodes one of the entries of the sequence, in a way that will be described later. We will denote the eigenstates using bra-ket notation in the form:
{displaystyle {|0rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle
and the corresponding eigenvalues as
{displaystyle \{{0},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{2},{1},{1},{2},{2},{3}.
We now take a unary operator, Uω, which acts as a subroutine that compares the different entries according to the search criteria. The algorithm does not specify how the subroutine works, but it must be a quantum subroutine working under a superposition of states. Moreover, it must act in a special way on one of the self states, |ω>, which will correspond to the input satisfying the search criterion. More precisely, we will require Uω with the following effects:
{ {displaystyle U_{\omega }||omega \rangle =-|omega \rangle }U_{\omega }|\omega \rangle =-|omega \rangle
{displaystyle U_{ "displaystyle" }| "xrangle" = "for all" { "for all" }U_{ "displaystyle" }| "xrangle" = "for all" { "xrangle" = "for all" }.
Specifically,
{displaystyle "angle" "angle" =1.
{displaystyle \langle \omega \xrangle =0 \langle x|\omega \rangle =0.
{displaystyle U_{ "\omega" =(I-2| "\omega" =2| "\rangle" =2| "\omega" =- "\omega" = "\omega". |\{ "\omega" =(I-2| "\omega" = -2| "\omega" =- "\omega" =- "\omega" =- "\omega".
{displaystyle U_ {\omega }{\xrangle = (I-2||memega \langle \memega |)|xrangle =|xrangle -2|memega \rangle \langle \memega |xrangle =|xrangle }U_{\omega }|xrangle =(I- 2|2|\omega \rangle \langle \omega |)|xrangle =|xrangle -2|\omega \rangle \langle \omega |xrangle =|xrangle =|xrangle .
Our goal is to identify the self-state |ω>, or equivalently, the eigenvalue ω, such that Uω acts specially on it.
The steps of Grover's algorithm are as follows:
Initialize the system to the state.
{ {{displaystyle ={{frac {1}{{sqrt {N}}}sum _{x}|xrangle}|{srangle ={frac {1}{sqrt {N}}}}}{sum _{x}|xrangle}|xrangle}}.
Perform the following iteration r (N) times. Where the function r (N) is described below.
Apply the operator { operator {displaystyle U_{\omega }}U_{\omega }}.
Apply the operator { {\displaystyle U_{s}=2\left|s\right angle s\right|-I}U_{s}=2\left|s\right angle s\right|-I}.
Perform the Ω measurement. The measure will correspond to the value λω with a certain probability that can be approximated to 1, for a certain N>>1. From λω, ω can be obtained.
We can write the operations performed:
{displaystyle s|s|rangle =1.
{displaystyle \langle s|somega =1. {displaystyle U_style \langle s|somega =2.
{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {N}}}}}{ "frac" }.
{displaystyle U_{s}(|source - {2}{\frac {2}{\sqrt {N}}|omega \rangle )=(2) (2) (|source -{2}{\sqrt {N}}|omega \rangle )}U_{s}(|source -{2}{\sqrt {N}}). { "2" {2}{ "2" - "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")
|{4} {4} {}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}
{ "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" + "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" - "disp
Summarizing, in a normal search of a data, if we have a disordered sequence, a linear inspection must be performed, which needs a time of O (N), so Grover's algorithm is a quite substantial improvement, avoiding, in addition, the need for prior sorting. The gain obtained is quadratic, which contrasts with other improvements of quantum algorithms that obtain exponential order improvements over their classical counterparts.
Like the previous quantum algorithm, Grover's algorithm is a probabilistic algorithm,
2.3 Brasard's algorithm. The time required to search for collisions in arbitrary functions r to 1: National Cryptfferent still.
The steps of Grover's algorithm are as follows:
Initialize the system to the state.
{ {{displaystyle ={{frac {1}{{sqrt {N}}}sum _{x}|xrangle}|{srangle ={frac {1}{sqrt {N}}}}}{sum _{x}|xrangle}|xrangle}}.
Perform the following iteration r (N) times. Where the function r (N) is described below.
Apply the operator { operator {displaystyle U_{\omega }}U_{\omega }}.
Apply the operator { {\displaystyle U_{s}=2\left|s\right angle s\right|-I}U_{s}=2\left|s\right angle s\right|-I}.
Perform the Ω measurement. The measure will correspond to the value λω with a certain probability that can be approximated to 1, for a certain N>>1. From λω, ω can be obtained.
We can write the operation performed:
{displaystyle s|s|rangle =1.
{displaystyle \langle s|somega =1. {displaystyle U_style \langle s|somega =2.
{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {N}}}}}{ "frac" }.
{displaystyle U_{s}(|source - {2}{\frac {2}{\sqrt {N}}|omega \rangle )=(2) (2) (|source -{2}{\sqrt {N}}|omega \rangle )}U_{s}(|source -{2}{\sqrt {N}}). { "2" {2}{ "2" - "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")
|{4} {4}
. \left|s\right|rangle \leftangle s\right|-I)(|s\rangle -{\frac {2}{\sqrt {N}}}}|omega \rangle )
{\frac {4}{\sqrt {N}}{\left {2}{\sqrt {N}}{\sqrt {N}}{\sqrt {N}|rangle - |{4} {4} {}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}
{ "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" + "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" =4} "displaystyle". 4}{N}}|rangle +{frac {2}{sqrt {N}}}|omega \rangle }=2|rangle - |{s "angle" - "angle" {4} "angle" {1} "angle" {1} "angle" + "angle" {2} "angle" {2} "angle" = "angle" {N-4} "angle" + "angle" {2} "angle" {2} "angle" {N}}}}|"angle
{1}{N{sqrt {N}}}}}left((N-4)\{sum _{xneq w}|xrangle +(3N-
After applying the two operators ( { {displaystyle U_{\omega }}U_{\omega }} and {displaystyle U_{s}}U_{s}} ), the amplitude of the searched element is increased. And this is a Grover iteration.
Summarizing, in a normal search of a data, if we have a disordered sequence, a linear inspection must be performed, which needs a time of O (N), so Grover's algorithm is a quite substantial improvement, avoiding, in addition, the need for prior sorting. The gain obtained is quadratic, which contrasts with other improvements of quantum algorithms that obtain exponential order improvements over their classical counterparts.
Like the previous quantum algorithm, Grover's algorithm is a probabilistic algorithm,
2.3 Brasard's algorithm. The time required to search for collisions in arbitrary functions r to 1: National Cryptologic Center Copyright (2016).
The three examples we have cited are problems with a clear, simple and unambiguous approach: find the factors of a number (Shor) or find an element in a list (Grover). In contrast, the problem we are interested in "designing new drugs by quantum computation" is not even posed in a concrete, non-generic way. Many paths could be considered and each of them would require a different algorithm:
The different avenues of attack have to be rethought and the corresponding algorithms would be much more different still.
We believe, in short, that it would be very interesting to be able to use the power of quantum computing in drug design".
Axiom of Virology.
That fundamental axiom states:
“All viruses on the planet follow the rule that in order to generate their genomes they have to make messenger RNA and be read by host ribosomes. ALL OF THEM.” ( Rancaniello, 2020).
Consequently, it is essential to develop an effective, inexpensive, orally administered antiviral with good bioavailability, selective toxicity and low cost.
There are 28 proteins on the surface of the virus SARS-CoV2. , but the 4 best known are: S,M,E, N.
.
Fig 3. Electrophysical forces
At the momento there is no effective antiviral treatment against this plague, except for three drugs very recently approved by the FDA:
(a) Molnupiravir (MK4482), a product of Merck & Ridgeback Therapeutics, which is an RNA analogue that is actually a pro-drug since it is transformed in the body into N-hydroxycytidine (NHC).
Indicated for patients with good oxygen saturation, with one or more risk factors. at a dose of 800 mg / 12 hours / 5 days, with an initial RR of 48% which later, as is often the case, was reduced to an RR of 30%.
This initial optimism is very characteristic of current pharmaceutical companies, with a tendency to inflate the data.
Molnupiravir is mutagenic: proven by the Ames test for Salmonella tiphymurium, the HPRT assay in hamster ovary cells and for developing teratogenicity in rats at high doses.
It was approved by the FDA by a narrow vote of 13 to 10.
b) Pfizer's Ritonavir + PF-07321332 combination. 100 mg of Ritonavir (which actually acts as an enzyme motivator9 + 300 mg of PF07321332 or Nirmatrelvir.
It is an Mpro (protease) inhibitor of SARS-CoV2. With a RR of 89% after 5 days of treatment and 85% after 5 days of treatment.
c) Monoclonal antibodies. That is, antibodies generated against a specific epitope of the virus surface. Epitope and antigenic determinant are the same, warning for computer scientists who are designing drugs. Leave the field free to virologists such as Prof. Luis Enjuanes, Mariano Esteban or Vicente Larraga, Isabel Sola, Sofía Zúñiga or Juan García Arriaza.
At the momento of review of this article mosto f Monoclonal antibodies are less active, only Barnmalizumab are useful right now.
Product Absolute risk NNT Teratogenicity
Molnupiravir (30%) 6,7 % 34 Yes6,7 %
FP07321332 (89%) 6,7 % 18 No
Monoclonal Ab (70-80%) 8% 11-25 No
Analyzing with biostatistical parameters, these are the differences.
Facts: Computers do not design ANYTHING if they are not well programmed to do so, but they are very useful and can be used for many purposes:
(a) Computational hit counting.
b) Detecting the three-dimensional arrangement of the minimal features in the form of an electron cloud mapping.
c) Resolution of searching for hydrophobic residues in molecules with Pm below 1500 (ideally between 350 and 900) - d) Interactions with the receptor.
d) Receptor interactions by calculating the binding energies (expressed in Kcal/mol) between these putative receptors for new (or repositioned) drugs, as shown in the figure.
e) Measurement of H (kcal/mol), S (cal/mol Kelvin) and Gibbs Energy G increments.
Short chapter of QM ( MD´s skip this one)
To give you an idea of the difficulties of quantum algorithms let us look at a couple of much simpler examples.
2.1 Shor's algorithm (1984) This algorithm is used to factor large integers.
Shor's theorem is from 1994, for factoring composite numbers.
Shor's algorithm consists of two parts:
Classical part.
We have a pseudo-random number a < N.
Compute the mcd(a, N). This can be done using Euclid's algorithm.
If the mcd(a, N) ≠ 1, then it is a nontrivial factor of N, so we are done.
Otherwise, we use the period-finding subprogram (see below) to find r, the period of the following function:
{displaystyle f(x)=a^{x}}f(x)=a^{x}}{mbox{mod}},
i.e. the smallest integer r for which
{displaystyle f(x+r)=f(x)}f(x+r)=f(x).
If r is odd, we go back to step 1.
If ar/2 ≡ -1 (mod N), we go back to step 1.
The factors of N are the mcd(ar/2 ± 1, N).
Quantum part: subprogram to find the period.
We start with a pair of input and output qubit registers with log2N qubits each, and initialize them to.
{ {{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xright\rangle }{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xright\rangle}\rangle|0right\rangle} }
Construct f(x) as a quantum function and apply it to the above state, to obtain.
{ {{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xrightrangle|f(x)\{rightrangle }{{displaystyle N^{-1/2}sum _{x=0}^{N-1}\left|xrightrangle|f(x)\{0}}.
We apply the quantum Fourier transform to the input register. The quantum Fourier transform at N points is defined as:
{{displaystyle U_{QFT}\left|x\rightrangle =N^{-1/2}sum _{y}e^{2\pi ixy/N}\left|y\rightrangle }U_{QFT}\left|x\rightrangle =N^{-1/2}sum _{y}e^{2\pi ixy/N}\left|y\rightrangle}.
Which leaves us in the following state:
{displaystyle N^{-1}sum _{x}sum _{y}e^{2\pi ixy/N}\left|yrightrangle|f(x)\N^{-1}sum _{x}sum _{y}e^{2\pi ixy/N}\left|yrightrangle|f(x)\N^{-1}sum _{x}sum _{y}e^{2\pi ixy/N}\left|yrightrangle|f(x)\N^{-1}rangle}.
We obtain a certain result y in the input register and f(x0) in the output register. This step is not necessary since, according to the deferred measurement principle, the result will be the same at the end of the algorithm regardless of whether a measurement is performed. However, for reasons of simplification in understanding the algorithm, we will include this step. Since f is periodic, the probability of measuring a certain y is given by
{{x:\,f(x)=f(x_{0})}e^{2\pi ixy/N}right|^{2}=N^{-1}\left|sum _{b}e^{2\pi i(x_{0}+rb)y/N}right|^{2}}N^{-1}\left|sum _{{x: \,f(x)=f(x_{0})}}e^{{2\pi ixy/N}}\right|^{2}=N^{{-1}}\left|\sum _{{b}}e^{{2\pi i(x_{0}+rb)y/N}}\right|^{2}
The analysis now shows that the higher this probability is, the more yr/N is close to an integer.
We convert y/N to an irreducible fraction, and extract the denominator r ', which is a candidate for r.
We check whether f(x) = f(x + r '). If so we are done.
If not, we obtain more candidates to r using values close to y, or multiples of r '. If any candidate satisfies the conditions, we are done.
If not, we go back to step 1 of the subprogram.
ummarized In the figure above. Factorization of n composite numbers.
Grover's algorithm is used to find a datum in an unordered list:
In other words, it tells us the time we would need for an exhaustive key test, it comes from Quantum mechanics It was invented by Lov K. Grover in 1996.
If an unordered sequence with N components is considered. The algorithm requires an N-dimensional state space H, which can be modeled with log2N qubits.
Let us number the entries of the sequence with 0, 1,.... (N-1); and let us select an observable Ω, acting on H, with N known distinct eigenvalues. Each of the eigenvalues of Ω encodes one of the entries of the sequence, in a way that will be described later. We will denote the eigenstates using bra-ket notation in the form:
{displaystyle {|0rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle ,|1rangle
and the corresponding eigenvalues as
{displaystyle \{{0},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{1},{2},{1},{1},{2},{2},{3}.
We now take a unary operator, Uω, which acts as a subroutine that compares the different entries according to the search criteria. The algorithm does not specify how the subroutine works, but it must be a quantum subroutine working under a superposition of states. Moreover, it must act in a special way on one of the self states, |ω>, which will correspond to the input satisfying the search criterion. More precisely, we will require Uω with the following effects:
{ {displaystyle U_{\omega }||omega \rangle =-|omega \rangle }U_{\omega }|\omega \rangle =-|omega \rangle
{displaystyle U_{ "displaystyle" }| "xrangle" = "for all" { "for all" }U_{ "displaystyle" }| "xrangle" = "for all" { "xrangle" = "for all" }.
Specifically,
{displaystyle "angle" "angle" =1.
{displaystyle \langle \omega \xrangle =0 \langle x|\omega \rangle =0.
{displaystyle U_{ "\omega" =(I-2| "\omega" =2| "\rangle" =2| "\omega" =- "\omega" = "\omega". |\{ "\omega" =(I-2| "\omega" = -2| "\omega" =- "\omega" =- "\omega" =- "\omega".
{displaystyle U_ {\omega }{\xrangle = (I-2||memega \langle \memega |)|xrangle =|xrangle -2|memega \rangle \langle \memega |xrangle =|xrangle }U_{\omega }|xrangle =(I- 2|2|\omega \rangle \langle \omega |)|xrangle =|xrangle -2|\omega \rangle \langle \omega |xrangle =|xrangle =|xrangle .
Our goal is to identify the self-state |ω>, or equivalently, the eigenvalue ω, such that Uω acts specially on it.
The steps of Grover's algorithm are as follows:
Initialize the system to the state.
{ {{displaystyle ={{frac {1}{{sqrt {N}}}sum _{x}|xrangle}|{srangle ={frac {1}{sqrt {N}}}}}{sum _{x}|xrangle}|xrangle}}.
Perform the following iteration r (N) times. Where the function r (N) is described below.
Apply the operator { operator {displaystyle U_{\omega }}U_{\omega }}.
Apply the operator { {\displaystyle U_{s}=2\left|s\right angle s\right|-I}U_{s}=2\left|s\right angle s\right|-I}.
Perform the Ω measurement. The measure will correspond to the value λω with a certain probability that can be approximated to 1, for a certain N>>1. From λω, ω can be obtained.
We can write the operations performed:
{displaystyle s|s|rangle =1.
{displaystyle \langle s|somega =1. {displaystyle U_style \langle s|somega =2.
{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {N}}}}}{ "frac" }.
{displaystyle U_{s}(|source - {2}{\frac {2}{\sqrt {N}}|omega \rangle )=(2) (2) (|source -{2}{\sqrt {N}}|omega \rangle )}U_{s}(|source -{2}{\sqrt {N}}). { "2" {2}{ "2" - "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")
|{4} {4} {}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}
{ "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" + "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" - "disp
Summarizing, in a normal search of a data, if we have a disordered sequence, a linear inspection must be performed, which needs a time of O (N), so Grover's algorithm is a quite substantial improvement, avoiding, in addition, the need for prior sorting. The gain obtained is quadratic, which contrasts with other improvements of quantum algorithms that obtain exponential order improvements over their classical counterparts.
Like the previous quantum algorithm, Grover's algorithm is a probabilistic algorithm,
2.3 Brasard's algorithm. The time required to search for collisions in arbitrary functions r to 1: National Cryptfferent still.
The steps of Grover's algorithm are as follows:
Initialize the system to the state.
{ {{displaystyle ={{frac {1}{{sqrt {N}}}sum _{x}|xrangle}|{srangle ={frac {1}{sqrt {N}}}}}{sum _{x}|xrangle}|xrangle}}.
Perform the following iteration r (N) times. Where the function r (N) is described below.
Apply the operator { operator {displaystyle U_{\omega }}U_{\omega }}.
Apply the operator { {\displaystyle U_{s}=2\left|s\right angle s\right|-I}U_{s}=2\left|s\right angle s\right|-I}.
Perform the Ω measurement. The measure will correspond to the value λω with a certain probability that can be approximated to 1, for a certain N>>1. From λω, ω can be obtained.
We can write the operation performed:
{displaystyle s|s|rangle =1.
{displaystyle \langle s|somega =1. {displaystyle U_style \langle s|somega =2.
{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {2}{ "frac" {N}}}}}{ "frac" }.
{displaystyle U_{s}(|source - {2}{\frac {2}{\sqrt {N}}|omega \rangle )=(2) (2) (|source -{2}{\sqrt {N}}|omega \rangle )}U_{s}(|source -{2}{\sqrt {N}}). { "2" {2}{ "2" - "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")(2 "2")
|{4} {4}
. \left|s\right|rangle \leftangle s\right|-I)(|s\rangle -{\frac {2}{\sqrt {N}}}}|omega \rangle )
{\frac {4}{\sqrt {N}}{\left {2}{\sqrt {N}}{\sqrt {N}}{\sqrt {N}|rangle - |{4} {4} {}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}}{}}}}
{ "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" + "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" - "displaystyle" - "displaystyle" =2|s "displaystyle" - "displaystyle" =4} "displaystyle". 4}{N}}|rangle +{frac {2}{sqrt {N}}}|omega \rangle }=2|rangle - |{s "angle" - "angle" {4} "angle" {1} "angle" {1} "angle" + "angle" {2} "angle" {2} "angle" = "angle" {N-4} "angle" + "angle" {2} "angle" {2} "angle" {N}}}}|"angle
{1}{N{sqrt {N}}}}}left((N-4)\{sum _{xneq w}|xrangle +(3N-
After applying the two operators ( { {displaystyle U_{\omega }}U_{\omega }} and {displaystyle U_{s}}U_{s}} ), the amplitude of the searched element is increased. And this is a Grover iteration.
Summarizing, in a normal search of a data, if we have a disordered sequence, a linear inspection must be performed, which needs a time of O (N), so Grover's algorithm is a quite substantial improvement, avoiding, in addition, the need for prior sorting. The gain obtained is quadratic, which contrasts with other improvements of quantum algorithms that obtain exponential order improvements over their classical counterparts.
Like the previous quantum algorithm, Grover's algorithm is a probabilistic algorithm,
2.3 Brasard's algorithm. The time required to search for collisions in arbitrary functions r to 1: National Cryptologic Center Copyright (2016).
The three examples we have cited are problems with a clear, simple and unambiguous approach: find the factors of a number (Shor) or find an element in a list (Grover). In contrast, the problem we are interested in "designing new drugs by quantum computation" is not even posed in a concrete, non-generic way. Many paths could be considered and each of them would require a different algorithm:
The different avenues of attack have to be rethought and the corresponding algorithms would be much more different still.
We believe, in short, that it would be very interesting to be able to use the power of quantum computing in drug design".
Axiom of Virology.
That fundamental axiom states:
“All viruses on the planet follow the rule that in order to generate their genomes they have to make messenger RNA and be read by host ribosomes. ALL OF THEM.” ( Rancaniello, 2020).
Consequently, it is essential to develop an effective, inexpensive, orally administered antiviral with good bioavailability, selective toxicity and low cost.
There are 28 proteins on the surface of the virus SARS-CoV2. , but the 4 best known are: S,M,E, N.
 Fig. 5 ( S (spike), N (nucleocapsid), M (membrane) and E (envelope) proteins. Taken from (16, 38, 39.)
At present, almost all the effort is focused on the 1274 amino acids of the protein S or spicule. As they are large structures (29, hydrophobic holes and unión to neuropilin receptors ( 40) which explaisn some of the clotting problems and tromboembolysm of SARs CoV2; , we can visualize them by cryo-EM and VR techniques and search for those hydrophobic residues formed by aromatic amino acids ("pockets") where receptors for molecules of molecular weight between 350 and 900. (17, 58)
The most appropriate therapeutic targets will be sought by relying on supercomputing and docking and screening of new molecules or repositioning of other drugs already accepted and authorized to treat other pathologies and orienting them to attack the therapeutic targets of SARS-CoV-2 (12, 18, 59, 66, 76)
Are computer-based drug designs useful? Yes, they are. Supercomputers do not design anything, they do not create anything if they are not well programmed to do so, but they are very useful as an accessory tool and we give two examples (55,60).
Fig. 4 Zanamavir (Relenza ®), designed with computational methods against the sialic receptor of the orthomyxovirus.
Or Indinavir, Crixivan®, directed against the HIV retrovirus protease.
Fig. 6.
Fig. 5 ( S (spike), N (nucleocapsid), M (membrane) and E (envelope) proteins. Taken from (16, 38, 39.)
At present, almost all the effort is focused on the 1274 amino acids of the protein S or spicule. As they are large structures (29, hydrophobic holes and unión to neuropilin receptors ( 40) which explaisn some of the clotting problems and tromboembolysm of SARs CoV2; , we can visualize them by cryo-EM and VR techniques and search for those hydrophobic residues formed by aromatic amino acids ("pockets") where receptors for molecules of molecular weight between 350 and 900. (17, 58)
The most appropriate therapeutic targets will be sought by relying on supercomputing and docking and screening of new molecules or repositioning of other drugs already accepted and authorized to treat other pathologies and orienting them to attack the therapeutic targets of SARS-CoV-2 (12, 18, 59, 66, 76)
Are computer-based drug designs useful? Yes, they are. Supercomputers do not design anything, they do not create anything if they are not well programmed to do so, but they are very useful as an accessory tool and we give two examples (55,60).
Fig. 4 Zanamavir (Relenza ®), designed with computational methods against the sialic receptor of the orthomyxovirus.
Or Indinavir, Crixivan®, directed against the HIV retrovirus protease.
Fig. 6.
 These are two valid examples to support that antiviral drugs can be designed with the help of docking techniques, screening and the use of supercomputing for the "in silico" screening of new anti viral drugs. (67)
Needless to say, no antiviral drug against SARS-CoV2 has been found so far (2). It is not easy. And history shows that some antivirals were discovered by serendipity, such as Amantadine, which improved the flu-like symptoms of a group of patients treated with antihistamines. Or the well-known Pfizer's Sildenafil (Viagra) ®, originally intended as an antihypertensive.
The aforementioned forces us to reconsider that this pandemic changes the paradigm and supercomputers and drug design and development programs are used by computational methods "in silico". And even a modification of the heuristic theory of docking is made and selective screening is performed on already approved drugs, in such a way that we can "save time" and find that pharmacological solution to the pandemic. Such would be the case of Raloxifene, a drug already authorized that has now been shown by pharmaceutical chemical calculation to have an inhibitory action on SAR-CoV 2 RNA polymerase. It should be pointed out that this is an aid to research, but supercomputers do not "design" drugs alone, they are only an accessory tool, they are not necessarily predictive (77) .
And perhaps most importantly: docking does not prove that the molecule actually binds to the receptor.
Docking or computerized molecular docking (1)
Virtual screenig or identification and search for compounds within a collection or set of similar (a library prior) and characteristic compounds. By means of computational hits we are experimenting. For example, in Pharmacognosy studies we identify all the alkaloids of ergot (Claviceps purpurea, which parasitizes rye (Secale cereale). This is virtual screening, using supercomputing or "in silico" modeling. We increase the probability of finding active molecules.
We need to actively screen for new antivirals in the hundreds and hundreds of as yet little known plants, marine tunicates, or the bowels of the earth.
Pharmacognosy is the science of searching for alkaloids and substances with active principles from the plant kingdom.
Of pharmacognosic origin we find other products. (6) Epsilon-Viniferin (-8.6 kcal/mol) or Dehirdectol (-10.3 kcal/mol), Carnosol (-8.2 kcal/mol), Arjunglucoside (-7.78 kcal/mol) and Rosmanol (-7.95 kcal/mol). (10)
These are two valid examples to support that antiviral drugs can be designed with the help of docking techniques, screening and the use of supercomputing for the "in silico" screening of new anti viral drugs. (67)
Needless to say, no antiviral drug against SARS-CoV2 has been found so far (2). It is not easy. And history shows that some antivirals were discovered by serendipity, such as Amantadine, which improved the flu-like symptoms of a group of patients treated with antihistamines. Or the well-known Pfizer's Sildenafil (Viagra) ®, originally intended as an antihypertensive.
The aforementioned forces us to reconsider that this pandemic changes the paradigm and supercomputers and drug design and development programs are used by computational methods "in silico". And even a modification of the heuristic theory of docking is made and selective screening is performed on already approved drugs, in such a way that we can "save time" and find that pharmacological solution to the pandemic. Such would be the case of Raloxifene, a drug already authorized that has now been shown by pharmaceutical chemical calculation to have an inhibitory action on SAR-CoV 2 RNA polymerase. It should be pointed out that this is an aid to research, but supercomputers do not "design" drugs alone, they are only an accessory tool, they are not necessarily predictive (77) .
And perhaps most importantly: docking does not prove that the molecule actually binds to the receptor.
Docking or computerized molecular docking (1)
Virtual screenig or identification and search for compounds within a collection or set of similar (a library prior) and characteristic compounds. By means of computational hits we are experimenting. For example, in Pharmacognosy studies we identify all the alkaloids of ergot (Claviceps purpurea, which parasitizes rye (Secale cereale). This is virtual screening, using supercomputing or "in silico" modeling. We increase the probability of finding active molecules.
We need to actively screen for new antivirals in the hundreds and hundreds of as yet little known plants, marine tunicates, or the bowels of the earth.
Pharmacognosy is the science of searching for alkaloids and substances with active principles from the plant kingdom.
Of pharmacognosic origin we find other products. (6) Epsilon-Viniferin (-8.6 kcal/mol) or Dehirdectol (-10.3 kcal/mol), Carnosol (-8.2 kcal/mol), Arjunglucoside (-7.78 kcal/mol) and Rosmanol (-7.95 kcal/mol). (10)
 In SARS CoV2 Mpro there have been far fewer mutations than in RBD so we understand that CoV Mpro targeted inhibitors could become broad spectrum antiviral drugs in case of COVID-19 and other CoV related diseases (Excakate4Covid project) We wrote this before the Pfizewr paxlovid product appeared, then we got it right. And by exposing the virus to various antivirals that act on different mechanisms, we managed to increase the error rate in the RNA polymerase of the virus artificially (catastrophic failures) ( 51) . And an association of a protease inhibitor + a polmerase inhibitor and a fusion protein inhibitor can be very useful in the clinic. But it is that before there were already crystal structure and docking results have shown that Ebselen inhibitors, TDZD-8 and α-ketoamide derivatives. Fig. 13. as potential drugs, since, in the laboratory we are looking for the concept of lethal mutation and entry into error catastrophes, which could be achieved with the application of several of these compounds in association, with which we could decrease their toxicity.
Pharmacophore modeling aims to detect the three-dimensional arrangement of the minimal features in the form of electron cloud mapping and, below is illustrated the pharmacophore model in which the interactions with the receptor are presented. The red sphere of high negative charge ( -2.19) and - 11.74. Pharmacophores.means that it can have electronic cloud pi, like benzene. (29,30)
Therapeutic targets for SARS-CoV-2 (32)
RNA polymerase. Example: Remdesivir, or Methysazone (3), for polypharmacology: Raloxifene.
polypharmacology: we already have other accepted molecules, but with another therapeutic indication, so we can use them for a new indication without having to carry out toxicological studies again
eEF1A (Eukaryotic translation elongation factor 1 alpha), in this case we do not really act on the virus, but on a host factor, but we have included it anyway. E.g.: Plitidepsin. Another example of library screening. This drug was used against multiple myeloma. Now it is being sought for Phase I application against Covid 19. (24, 44, 45)
The structure of Mpro Examples would be Ebselen also called PZ51 DR3305, or SPI-1005, Carfilzomib (7,8,9, 25, 31).
We are going to focus on this Mpro, using pharmaceutical chemical calculation techniques looking for molecules of molecular weight between 340 and 900 that bind to these hydrophobic residues that form in certain regions and test on a "brute force" basis various drugs that have shown activity on Mpro are Etoposide, Hesperidin, Velpatasvir, Diosmin and Venetotoclax.
RnRp or mRNA polymerase-dependent Ribonuclease (4). It was discovered from studies on Mengovirus and polymyelitis virus when it was observed that these viruses were insensitive to actinomycin D, a drug that inhibits DNA-catalyzed cellular RNA synthesis. This lack of sensitivity suggested that there is a virus-specific enzyme an enzyme specific to RNA viruses that catalyzes RNA replication from an RNA template. that could copy RNA from an RNA template rather than from a DNA template.( 41, 48, 49)
Like MK-7376, they appear to have actions as inhibitors of protein acidic endonuclease function or the two compounds CoViTris2020 and ChloViD2020 (5) or Mr224 (14).
Ionophore channels: e.g. 0.6% Ivermectin. This "in vitro" action is also being studied "in vivo" in clinical trials. (48, 68)
Alpha-keto glutarase protease inhibitors. E.g. Telaprevir, Borceprevir. These drugs inhibit Hepatitis C virus protease, specifically its HCV serine NS3-4A PI, an enzyme essential for virus replication. Doubtful that they have any action against SARS-CoV-2, although both are Baltimore classification IV single-catenary RNA viruses.
Elevation of endoplasmic pH, inhibiting viral metabolism: Hydroxychloroquine.
Chloroquine and its hydroxy derivative would lead us to the Surgisphere scandal and The Lancet, and we prefer to leave it aside. Chloroquine has several mechanisms of affecting SARS-CoV-2, preventing its entry and diffusion into the cell, but the increase in intracellular Ph is perhaps the most important.(41) Cysteine protease inhibitors also plays a role . Other protein inhibitors ( 52) also plays a relevant role in filovirus entry.
Fusion Inhibitors. They inhibit the processes of anchoring and fusion. Specifically to the TMPRSS2 protein.(21, 41, 46, 49)
Here we would have Camustat acetate (53, 55) , which is undergoing clinical trials. Drugs that inhibit the anchoring and fusion processes. Specifically the TMPRSS2 protein. (41, 45) and also other protein inhibase drugs ( 58).
When the SARS-CoV2 virus anchors to the ACE/2 h receptor of epithelial cells, it undergoes two cleavages.(21, 22, 46, 69, 72,77).
The role of the 4 polybase aminoacides and PCF (60) are for some Scientific such as David Baltimore , the “Smoking gun” of the non zoonosis origin of the virus.
1) One is furin-dependent, a protein existing in the host's own system, but which the virus, laddily uses against the host (the host, as they call it in South American Virology books), which it does to take advantage of our system to start generating stable meta virons by anchoring (S1) and then fusing (S2). (23)
These are very important steps on which it is convenient to deepen, as we will do later.
2) The second cleavage occurs with S2 changing conformation and a fusion protein appears, absolutely necessary to penetrate the human epithelial cell that is the host (the possibility has not been demonstrated in endothelial cells, as proposed by two American thoracic surgeons.
There is a conformational change and we go from a pre-fusion PDB-6VSB protein to another post-fusion protein. PDB:6LXT. ( 33, 34, 35, 36, 37)
Here Camustat Mesylate acts, inhibiting TMPRSS2 and on which clinical trials are underway to determine its use in the clinic. This virus fusion protein is critical, along with the anchor protein.(19)
When does cleavage occur?
On the cell surface or in the endoplasm?
Is this process of conformational change influenced by pH?
A very basic pH (achieved for example by taking HCQ, raising the intracellular pH above 8.5 would inhibit the endonuclear processes of the virus, producing its reproductive slowing and inhibiting the process of cleavage.
The "cleavage" (anglicism to define enzymatic cleavage) of S1 and S2 is performed by the furin cleavage protein call. Yes, the same furin cleavage proteins which, according to the Chinese dissident, are one of the three characteristics of the virus itself to have been synthesized in a laboratory, from two common cold corona viruses and then an insertion of the spicule by techniques that would have taken about six months. This would also explain the notable differences between the three proteins of the horseshoe bat, pangolin and SARS-CoV 2 spike.
Therefore, antiviral drugs may have different "targets", but they all ultimately go through the caudine forks of having to translate their m RNA into proteins or genomic material using the organelles, ribosomes and enzymes of the host they infect.
The most appropriate therapeutic targets will be sought by relying on supercomputing and docking/writing of new molecules or repositioning of other drugs already accepted and authorized to treat other pathologies.
Supercomputing is not the same as using quantum algorithms (parallel computing; quantum computing requires special algorithms for each type of problem).
In SARS CoV2 Mpro there have been far fewer mutations than in RBD so we understand that CoV Mpro targeted inhibitors could become broad spectrum antiviral drugs in case of COVID-19 and other CoV related diseases (Excakate4Covid project) We wrote this before the Pfizewr paxlovid product appeared, then we got it right. And by exposing the virus to various antivirals that act on different mechanisms, we managed to increase the error rate in the RNA polymerase of the virus artificially (catastrophic failures) ( 51) . And an association of a protease inhibitor + a polmerase inhibitor and a fusion protein inhibitor can be very useful in the clinic. But it is that before there were already crystal structure and docking results have shown that Ebselen inhibitors, TDZD-8 and α-ketoamide derivatives. Fig. 13. as potential drugs, since, in the laboratory we are looking for the concept of lethal mutation and entry into error catastrophes, which could be achieved with the application of several of these compounds in association, with which we could decrease their toxicity.
Pharmacophore modeling aims to detect the three-dimensional arrangement of the minimal features in the form of electron cloud mapping and, below is illustrated the pharmacophore model in which the interactions with the receptor are presented. The red sphere of high negative charge ( -2.19) and - 11.74. Pharmacophores.means that it can have electronic cloud pi, like benzene. (29,30)
Therapeutic targets for SARS-CoV-2 (32)
RNA polymerase. Example: Remdesivir, or Methysazone (3), for polypharmacology: Raloxifene.
polypharmacology: we already have other accepted molecules, but with another therapeutic indication, so we can use them for a new indication without having to carry out toxicological studies again
eEF1A (Eukaryotic translation elongation factor 1 alpha), in this case we do not really act on the virus, but on a host factor, but we have included it anyway. E.g.: Plitidepsin. Another example of library screening. This drug was used against multiple myeloma. Now it is being sought for Phase I application against Covid 19. (24, 44, 45)
The structure of Mpro Examples would be Ebselen also called PZ51 DR3305, or SPI-1005, Carfilzomib (7,8,9, 25, 31).
We are going to focus on this Mpro, using pharmaceutical chemical calculation techniques looking for molecules of molecular weight between 340 and 900 that bind to these hydrophobic residues that form in certain regions and test on a "brute force" basis various drugs that have shown activity on Mpro are Etoposide, Hesperidin, Velpatasvir, Diosmin and Venetotoclax.
RnRp or mRNA polymerase-dependent Ribonuclease (4). It was discovered from studies on Mengovirus and polymyelitis virus when it was observed that these viruses were insensitive to actinomycin D, a drug that inhibits DNA-catalyzed cellular RNA synthesis. This lack of sensitivity suggested that there is a virus-specific enzyme an enzyme specific to RNA viruses that catalyzes RNA replication from an RNA template. that could copy RNA from an RNA template rather than from a DNA template.( 41, 48, 49)
Like MK-7376, they appear to have actions as inhibitors of protein acidic endonuclease function or the two compounds CoViTris2020 and ChloViD2020 (5) or Mr224 (14).
Ionophore channels: e.g. 0.6% Ivermectin. This "in vitro" action is also being studied "in vivo" in clinical trials. (48, 68)
Alpha-keto glutarase protease inhibitors. E.g. Telaprevir, Borceprevir. These drugs inhibit Hepatitis C virus protease, specifically its HCV serine NS3-4A PI, an enzyme essential for virus replication. Doubtful that they have any action against SARS-CoV-2, although both are Baltimore classification IV single-catenary RNA viruses.
Elevation of endoplasmic pH, inhibiting viral metabolism: Hydroxychloroquine.
Chloroquine and its hydroxy derivative would lead us to the Surgisphere scandal and The Lancet, and we prefer to leave it aside. Chloroquine has several mechanisms of affecting SARS-CoV-2, preventing its entry and diffusion into the cell, but the increase in intracellular Ph is perhaps the most important.(41) Cysteine protease inhibitors also plays a role . Other protein inhibitors ( 52) also plays a relevant role in filovirus entry.
Fusion Inhibitors. They inhibit the processes of anchoring and fusion. Specifically to the TMPRSS2 protein.(21, 41, 46, 49)
Here we would have Camustat acetate (53, 55) , which is undergoing clinical trials. Drugs that inhibit the anchoring and fusion processes. Specifically the TMPRSS2 protein. (41, 45) and also other protein inhibase drugs ( 58).
When the SARS-CoV2 virus anchors to the ACE/2 h receptor of epithelial cells, it undergoes two cleavages.(21, 22, 46, 69, 72,77).
The role of the 4 polybase aminoacides and PCF (60) are for some Scientific such as David Baltimore , the “Smoking gun” of the non zoonosis origin of the virus.
1) One is furin-dependent, a protein existing in the host's own system, but which the virus, laddily uses against the host (the host, as they call it in South American Virology books), which it does to take advantage of our system to start generating stable meta virons by anchoring (S1) and then fusing (S2). (23)
These are very important steps on which it is convenient to deepen, as we will do later.
2) The second cleavage occurs with S2 changing conformation and a fusion protein appears, absolutely necessary to penetrate the human epithelial cell that is the host (the possibility has not been demonstrated in endothelial cells, as proposed by two American thoracic surgeons.
There is a conformational change and we go from a pre-fusion PDB-6VSB protein to another post-fusion protein. PDB:6LXT. ( 33, 34, 35, 36, 37)
Here Camustat Mesylate acts, inhibiting TMPRSS2 and on which clinical trials are underway to determine its use in the clinic. This virus fusion protein is critical, along with the anchor protein.(19)
When does cleavage occur?
On the cell surface or in the endoplasm?
Is this process of conformational change influenced by pH?
A very basic pH (achieved for example by taking HCQ, raising the intracellular pH above 8.5 would inhibit the endonuclear processes of the virus, producing its reproductive slowing and inhibiting the process of cleavage.
The "cleavage" (anglicism to define enzymatic cleavage) of S1 and S2 is performed by the furin cleavage protein call. Yes, the same furin cleavage proteins which, according to the Chinese dissident, are one of the three characteristics of the virus itself to have been synthesized in a laboratory, from two common cold corona viruses and then an insertion of the spicule by techniques that would have taken about six months. This would also explain the notable differences between the three proteins of the horseshoe bat, pangolin and SARS-CoV 2 spike.
Therefore, antiviral drugs may have different "targets", but they all ultimately go through the caudine forks of having to translate their m RNA into proteins or genomic material using the organelles, ribosomes and enzymes of the host they infect.
The most appropriate therapeutic targets will be sought by relying on supercomputing and docking/writing of new molecules or repositioning of other drugs already accepted and authorized to treat other pathologies.
Supercomputing is not the same as using quantum algorithms (parallel computing; quantum computing requires special algorithms for each type of problem).
 MATERIAL AND METHODS.
We will use the programs referenced below and the so-called Glide-Schodinger dock score, which is nothing else than the EFEB (estimated free energy of binding). The docking techniques will allow us to find a specific antiviral against SARS-CoV-2, that "magic bullet" as Ehrlich said.
We would use the GlideScore programs (84)
a) The GS calculates EFEB empirically and using a heuristic approximation of the free energy by measuring ligand forces, and they have other subcomponents such as calculation of electrostatic forces, van deer Waals, and force fields. The GlideScore is used for virtual screening (85), expressed in kilocalories/mole and the more negative the value, the stronger the binding. Two subprograms called XP GlideScore and SP / HTVS GlideScore.
b) Emodel has a more significant weighting of the force field components (electrostatic and van der Waals energies), which makes it very suitable for comparing conformers, but much less suitable for comparing chemically distinct species. Therefore, Glide uses Emodel to choose the "best" pose of a ligand (pose selection) and then ranks these best positions.
c) Glide for docking uses a large class of computer algorithms that attempt to find an optimal location of a rigid or flexible ligand at the receptor binding site. Ligands are typically small molecule; peptide-protein and protein-protein docking algorithms are currently in active development. The docking algorithms also generate a score that attempts to distinguish between molecules that bind tightly at their optimal location.
d) ChemOffice+ Cloud is a robust and comprehensive suite specifically designed to simplify, facilitate and accelerate chemical communication. The cloud-native chemistry communication suite and adds access to a powerful set of tools to enable scientific research. Performs fast structure searches on all documents where chemical information is stored.
3D
e) Chimera, a 145 Mb UCSF program, free to use, not for design, but for visualization.
f) Schrödinger's Firm Programs
You use the PDB file name stored in the RCSB protein data bank and an SDF rn rl file with nearly a million lead-like compounds from a database such as ZINC in a docking program, and sit back while the computer docks each compound to each binding pocket in the protein. In reality, meaningful docking calculations require careful preparation of the receptor and ligand structures before the docking programs can do their work. With the Schrödinger suite of programs, most of the receptor preparation is done with the Protein Preparation Wizard, while ligand preparation is done with the Ligand Preparation Wizard.
g) Master Molecule Editing Program.
h) OTHERS: Autodock, EKLUSTAR, MODELLER, Chem sketch, bin RalMol, Jmol, VMD Cn-d, nanome.
CONCLUSIONS.
At present the treatment of COVID 19 is complex and multidisciplinary. It requires coordination between emergency services, intensivists and internal medicine specialists.
Drugs such as Molnupiravir or paxlovid are very interesting options, but they are not the only ones and supercomputing should continue to be used to search for specific antiviral drugs that do not generate resistance.
The global fight against this pandemic will require the efforts of both research and improvement of vaccines, as well as the development of new antivirals.(70, 71, 73, 75).
The pure Darwinian evolutionary mechanism of adaptation to the host will very possibly lead to the appearance of new escape variants, as has happened in other viruses, including DNA viruses, which mutate much less (e.g. hepatitis B), so relying solely and exclusively on mRNA vaccines seems to us to be a mistake. And we will have to have contingency plans prepared for this. We believe that screening programs for new drugs will be cost-effective and are a useful therapeutic tool to control the disease in its early stages (even subclinical) and help control cases.
We will not be able to consider the pandemic under control until the cumulative incidence is below 1 case per 100,000 inhabitants and we are far from that at the moment and this situation will last at least three more years.
The need to implement new antiviral drugs is paramount to stop the pandemic at this stage.
REFERENCES
(1)José L.Medina-Franco Jl, Fernández-de Gortaria E, Naveja J.Avances en el diseño de fármacos asistido por computadora. Progress in computer-aided drug design. Educación Química; 2015 26, (3) 180-186.
(2)Cava C, Bertoli G, Castiglioni I. In Silico Discovery of Candidate Drugs against Covid-19. Viruses. 2020 Apr 6;12(4):404. doi: 10.3390/v12040404. PMID: 32268515; PMCID: PMC7232366.
(3)Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020;252:117652. doi:10.1016/j.lfs.2020.117652
(4)Koonin, E.V., Gorbalenya, A.E. and Chumakov, K.M.(1989), Tentative identification of RNA-dependent RNA polymerases of dsRNA viruses and their relationship to positive strand RNA viral polymerases, FEBS Letters, 252, doi: 10.1016/0014-5793(89)80886-5
(5)Rabie AM. CoViTris2020 and ChloViD2020: a striking new hope in COVID-19 therapy. Mol Divers. 2021 Jan 3:1–16. doi: 10.1007/s11030-020-10169-0. Epub ahead of print. PMID: 33389560; PMCID: PMC7778709.
(6) Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020;252:117652. doi:10.1016/j.lfs.2020.117652
(7)Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020;252:117652. doi:10.1016/j.lfs.2020.117652
(8)Alnajjar R, Mostafa A, Kandeil A, Al-Karmalawy AA. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon. 2020 Dec;6(12):e05641. doi: 10.1016/j.heliyon.2020.e05641. Epub 2020 Dec 3. PMID: 33294721; PMCID: PMC7713577.
(9)Wu J , Wang H , Li B . Structure-aided ACEI-capped remdesivir-loaded novel PLGA nanoparticles: toward a computational simulation design for anti-SARS-CoV-2 therapy. Phys Chem Chem Phys. 2020 Dec 23;22(48):28434-28439. doi: 10.1039/d0cp04389c. PMID: 33305304.
(10)Keretsu, S., Bhujbal, S.P. & Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci Rep 10, 17716 (2020). https://doi.org/10.1038/s41598-020-74468-
(11)Zhenming Jin, Xiaoyu Du, Yechun Xu, Yongqiang Deng, Meiqin Liu, Yao Zhao, Bing Zhang, Xiaofeng Li, Leike Zhang, Yinkai Duan, Jing Yu, Lin Wang, Kailin Yang, Fengjiang Liu, Tian You, Xiaoce Liu, Xiuna Yang, Fang Bai, Hong Liu, Xiang Liu, Luke W. Guddat, Gengfu Xiao, Chengfeng Qin, Zhengli Shi, Hualiang Jiang, Zihe Rao, Haitao YangbioRxiv 2020.02.26.964882; doi: https://doi.org/10.1101/2020.02.26.964882
(12) Mohapatra S, Nath P, Chatterjee M, Das N, Kalita D, et al. (2020) Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLOS ONE 15(11): e0241543. https://doi.org/10.1371/journal.pone.0241543
(13)Mohapatra S, Nath P, Chatterjee M, Das N, Kalita D, et al. (2020) Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLOS ONE 15(11): e0241543. https://doi.org/10.1371/journal.pone.0241543
(14)El Hassab Mahmoud A., Shoun Aly A., Al-Rashood T., Al-Warhi Tarfah, Identification of a New Potential SARS-COV-2 RNA-Dependent RNA Polymerase Inhibitor via Combining Fragment-Based Drug Design, Docking, Molecular Dynamics, and MM-PBSA Calculations JFrontiers in Chemistry.2020; 8:915-916. DOI=10.3389/fchem.2020.584894
(15)K. Abraham Peele, Chandrasai Potla Durthi, T. Srihansa, S. Krupanidhi, Vijaya Sai Ayyagari, D. John Babu, M. Indira, A. Ranganadha Reddy, T.C. Venkateswarulu,Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study,Informatics in Medicine Unlocked; 2020,(19)100-345ISSN 2352-9148,https://doi.org/10.1016/j.imu.2020.100345.
(16) Giwa AL, Desai A, Duca A. Novel 2019 coronavirus SARS-CoV-2 (COVID-19): An updated overview for emergency clinicians. Emerg Med Pract. 2020 May 1;22(5):1-28. Epub 2020 Mar 24. PMID: 32207910. ( figura de las 4 proteinas9
(17) Peele KA, Potla Durthi C, Srihansa T, Krupanidhi S, Ayyagari VS, Babu DJ, Indira M, Reddy AR, Venkateswarulu TC. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform Med Unlocked. 2020;19:100345. doi: 10.1016/j.imu.2020.100345. Epub 2020 May 11. PMID: 32395606; PMCID: PMC7211761.
(18) Li F. Structure, function, and evolution of coronavirus spike proteins. Annual Rev. Virol. 2016; 3(1):237‐261.
(19)Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. China Life Sci. 2020; 63(3):457‐460.
(20) Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. 2020;579(7798):270‐273.
(21)Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Science Alliance. 2020;3(9):e202000786.
(22)Hoffmann M, Hofmann‐Winkler H, Smith JC, et al. Camostat mesylate inhibits SARS‐CoV‐2 activation by TMPRSS2‐related proteases and its metabolite GBPA exerts antiviral activity. bioRxiv 2020.
(23)Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik O, Rohde C, Klenk HD, Garten W, Steinmetzer T, Böttcher-Friebertshäuser E. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. 2020 Jul 23;3(9):e202000786. doi: 10.26508/lsa.202000786. PMID: 32703818; PMCID: PMC7383062.
(24)Walls AC, Tortorici MA, Bosch B‐J, et al. Cryo‐electron microscopy structure of a coronavirus spike glycoprotein trimer. 2016; 531(7592): 114‐ 117.
(25) Gomes CP, Fernandes DE, Casimiro F, da Mata GF, Passos MT, Varela P, Mastroianni-Kirsztajn G, Pesquero JB. Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front Cell Infect Microbiol. 2020 Dec 8;10:589505. doi: 10.3389/fcimb.2020.589505. PMID: 33364201; PMCID: PMC7753008.
(26)Breining P, Frølund AL, Højen JF, Gunst JD, Staerke NB, Saedder E, Cases-Thomas M, Little P, Nielsen LP, Søgaard OS, Kjolby M. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin Pharmacol Toxicol. 2021 Feb;128(2):204-212. doi: 10.1111/bcpt.13533. Epub 2020 Nov 22. PMID: 33176395.
(27)Hydroxychloroquine and Azithromycin as a treatment of COVID-19: preliminary results of an open-label non-randomized clinical trial medRxiv (2020) doi: 10.1101/2020.03.16.20037135International Journal of Antimicrobial Agents (2020) doi: 10.1016/j.ijantimicag.2020.105949
(28)Heidi Ledford, Richard Van Noorden, «High-profile coronavirus retractions raise concerns about data oversight,» Nature (05 Jun 2020), doi: https://doi.org/10.1038/d41586-020-01695-w
(29)Prinz JHWu HSarich Met al.Markov models of molecular kinetics: generation and validation.J Chem Phys. 2011; 134174105
(30) Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein Alexandra C. Walls, Young-Jun Park, M. Alexandra Tortorici, Abigail Wall, Andrew T. McGuire, David VeeslerbioRxiv 2020.02.19.956581; doi: https://doi.org/10.1101/2020.02.19.956581
(31)Cryo-EM structure of S-Trimer, a subunit vaccine candidate for COVID-19Jiahao Ma, Danmei Su, Xueqin Huang, Ying Liang, Yan Ma, Peng Liang, Sanduo ZhengbioRxiv 2020.09.21.306357; Rxiv preprint 306357 (21 Sep 2020) https://doi.org/10.1101/2020.09.21.306357
(32)Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, Liu X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J Med Chem. 2020 Nov 12; 63(21):12256-12274. doi: 10.1021/acs.jmedchem.0c00502. Epub 2020 Jun 25. PMID: 32539378; PMCID: PMC7315836.
(33)Lan, J., Ge, J., Yu, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature (2020). https://doi.org/10.1038/s41586-020-2180-5).
(34)Shang, J., Ye, G., Shi, K. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020). https://doi.org/10.1038/s41586-020-2179-y
(35) Lan, J., Ge, J., Yu, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020). https://doi.org/10.1038/s41586-020-2180-5
(36)Shang, J., Ye, G., Shi, K. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020). https://doi.org/10.1038/s41586-020-2179-y
(37) Wenfei Song, Miao Gui, Ye Xiang, «Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2, » PLoS Pathogens, 14: e1007236 (13 Aug 2018), doi: https://doi.org/10.1371/journal.ppat.1007236.
(38)Ludovico Cantuti-Castelvetri, Ravi Ojha, Liliana D. Pedro, Minou Djannatian, Jonas Franz, Suvi Kuivanen, Katri Kallio, Tuğberk Kaya, Maria Anastasina, Teemu Smura, Lev Levanov, Leonora Szirovicza, Allan Tobi, Hannimari Kallio-Kokko, Pamela Österlund, Merja Joensuu, Frédéric A. Meunier, Sarah Butcher, Martin Sebastian Winkler, Brit Mollenhauer, Ari Helenius, (39)Ozgun Gokce, Tambet Teesalu, Jussi Hepojoki, Olli Vapalahti, Christine Stadelmann, Giuseppe Balistreri, Mikael Simons NRP1 Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system bioRxiv 2020.06.07.137802; doi: https://doi.org/10.1101/2020.06.07.137802
(40) Neuropilin-1 is a host factor for SARS-CoV-2 infectionJames L. Daly, Boris Simonetti, Carlos Antón-Plágaro, Maia Kavanagh Williamson, Deborah K. Shoemark, Lorena Simón-Gracia, Katja Klein, Michael Bauer, Reka Hollandi, Urs F. Greber, Peter Horvath, Richard B. Sessions, Ari Helenius, Julian A. Hiscox, Tambet Teesalu, David A. Matthews, Andrew D. Davidson, Peter J. Cullen, Yohei YamauchibioRxiv 2020.06.05.134114; doi: https://doi.org/10.1101/2020.06.05.134114
(41)Padmanabhan, Pranesh; Desikan, Rajat; Dixit, Narendra (2020): Targeting TMPRSS2 and Cathepsin B/L Together May Be Synergistic Against SARS-CoV-2 Infection. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.12213125.v2
(42)Ruoslahti «RGD and other recognition sequences for integrins» Annu. Rev. Cell Dev. Biol. 1996, 12: 697–715».
(43)Friess H, Kleeff J, Isenmann R, Malfertheiner P, Büchler MW. Adaptation of the human pancreas to inhibition of luminal proteolytic activity. Gastroenterology. 1998; 115(2): 388‐ 396.
(44)Ohkoshi M, Oka T. Clinical experience with a protease inhibitor [N, N‐dimethylcarbamoylmethyl 4‐ (4‐guanidinobenzoyloxy) ‐phenylacetate] methanesulfate for prevention of recurrence of carcinoma of the mouth and in treatment of terminal carcinoma. J. Maxillofacial Surg. 1984; 12(4): 148‐ 152.
(45)Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015; 116: 76‐ 84.
(46)Shirato K, Kawase M, Matsuyama S. Middle east respiratory syndrome coronavirus infection mediated by the trans membrane serine protease TMPRSS2. J Virol. 2013; 87(23): 12552‐ 12561.
(47)Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012; 86(12): 6537‐ 6545.
(48)Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. Protease‐mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci. 2005; 102(35): 12543‐ 12547.
(49)Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012; 86(12): 6537‐ 6545.
(50)Reagan‐Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659‐661.
(51)Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP‐mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875‐894.
(52)Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76‐84.
(53)Midgley I, Hood AJ, Proctor P, et al. Metabolic fate of 14C‐camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica. 1994; 24(1): 79‐ 92.
(54) Ohki SNishiyama H Ozeki KIto HHirata F.Studies on absorption, distribution, metabolism and excretion of [14C] FOY-305.Gendai-Iryo. 1980; 12: 71-82
(55)Senokuchi K, Nakai H, Nakayama Yet al. New orally active serine protease inhibitors.J Med Chem. 1995; 38: 2521-2523
(56) Beckh KGoke BMuller RArnold R.Elimination of the low-molecular weight proteinase inhibitor camostate (FOY 305) and its degradation products by the rat liver.Res Exp Med (Berl). 1987; 187: 401-406
(57) Beckh KWeidenbach HWeidenbach FMuller RAdler G.Hepatic and pancreatic metabolism and biliary excretion of the protease inhibitor camostat mesilate.Int J Pancreatol. 1991; 10: 197-205
(58) Senokuchi KNakai HNakayama Yet al.New orally active serine protease inhibitors.J Med Chem. 1995; 38: 2521-2523
(59)Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020; 181(2): 271‐ 280.e8.
(60)Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Science Alliance. 2020; 3(9):e202000786.
(61)Yamaya M, Shimotai Y, Hatachi Y, et al. The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm Pharmacol Ther. 2015; 33: 66‐ 74.
(62)Hempel T Raich L Olsson Set al. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat.Chemical Science. 2021;
(63)Midgley I, Hood AJ, Proctor P, et al. Metabolic fate of 14C‐camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica. 1994; 24(1): 79‐ 92.
(64)Sun W, Zhang X, Cummings MD, et al. Targeting enteropeptidase with reversible covalent inhibitors to achieve metabolic benefits. Journal of Pharmacology and Experimental Therapeutics. 2020; JPET– AR. http://dx.doi.org/10.1124/jpet.120.000219.
(65) Spraggon G, Hornsby M, Shipway A, et al. Active site conformational changes of prostasin provides a new mechanism of protease regulation by divalent cations. Protein Sci. 2009; 18(5): 1081‐ 1094.
(66) Hempel T, Raich L, Olsson S, et al. Molecular mechanism of SARS‐CoV‐2 cell entry inhibition via TMPRSS2 by Camostat and Nafamostat mesylate. bioRxiv 2020:2020.07.21.214098 https://doi.org/10.1101/2020.07.21.214098
(67) Depfenhart M, de Villiers D, Lemperle G, Meyer M, Di Somma S. Potential new treatment strategies for COVID‐19: is there a role for bromhexine as add‐on therapy? Intern Emerg Med. 2020; 15(5): 801‐ 812.
(68)13/20 Informationen der Institutionen und Behörden: BMG: Zentrale Beschaffung vonArzneimitteln zur Therapie schwerwiegender Verläufe COVID-19 infizierter Patienten undVerteilung an Apotheken durch die Bundeswehr (24.03.2020). 2020 [cited 7/9/2020]; Availablefrom: https://www.abda.de/fuer-apotheker/arzneimittelkommission/amk-nachrichten/detail/13-20-informationen-der-institutionen-und-behoerden-bmg-zentrale-beschaffung-von-arzneimittelnzur-therapie-schwerwiegender-verlaeufe-covid-19-infizierter-patienten-und-verteilung-anapotheken-durch-die-bundeswehr/
(69)Hofmann-Winkler H, Moerer O, Alt-Epping S, Bräuer A, Büttner B, Müller M, et al. CamostatMesylate but Not Hydroxychloroquine May Reduce Severity of Organ Failure in COVID-19Sepsis: A Case-Control Study (6/15/2020) 2020
(70)Ji W, Huh K, Kang M, Hong J, Bae GH, Lee R, et al. Effect of Underlying Comorbidities on theInfection and Severity of COVID-19 in Korea: a Nationwide Case-Control Study. J Korean MedSci. 2020 Jun 29;35(25):e237.
(71)Yamamoto M, Kiso M, Sakai-Tagawa Y, Iwatsuki-Horimoto K, Imai M, Takeda M, Kinoshita N, Ohmagari N, Gohda J, Semba K, Matsuda Z, Kawaguchi Y, Kawaoka Y, Inoue JI. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses. 2020 Jun 10;12(6):629. doi: 10.3390/v12060629. PMID: 32532094; PMCID: PMC7354595.
(72)Ou T, Mou H, Zhang L, Ojha A, Choe H, Farzan M (2021) Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog 17(1): e1009212. doi:10.1371/journal.ppat.1009212
(73)Maurizio Guastalegname, Alfredo Vallone, Could Chloroquine /Hydroxychloroquine Be Harmful in Coronavirus Disease 2019 (COVID-19) Treatment?, Clinical Infectious Diseases, Volume 71, Issue 15, 1 August 2020, Pages 888–889, https://doi.org/10.1093/cid/ciaa321
(74)Siyu Xiu, Alexej Dick, Han Ju, Sako Mirzaie, Fatemeh Abdi, Simon Cocklin, Peng Zhan, and Xinyong Liu Inhibitors of SARS-CoV-2 Entry: Current and Future OpportunitiesJournal of Medicinal Chemistry 2020 63 (21), 12256-12274DOI: 10.1021/acs.jmedchem.0c00502
https://www.bbc.com/mundo/noticias-internacional-55406204
(75)Song W, Gui M, Wang X, Xiang Y (2018) Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 14(8): e1007236. https://doi.org/10.1371/journal.ppat.1007236
(76)Hoffmann et al., Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and itsmetabolite GBPA exerts antiviral activity, EBioMedicine (2021), https://doi.org/10.1016/j.ebiom.2021.103255
(77)Barnard DL, Day CW, Bailey K, et al. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir Chem Chemother 2006; 17:275–84.
https://www.clinicaltrials.gov/ct2/show/NCT04381936
https://www.clinicaltrialsregister.eu/
(https://www.clinicaltrials.gov/ct2/show/NCT04381936
https://www.clinicaltrials.gov/ct2/show/NCT04321616
https://trialsitenews.com/study-combines-korean-pancreatitisdrug-foistar-with-
https://www.medrxiv.org/content/10.1101/2
MATERIAL AND METHODS.
We will use the programs referenced below and the so-called Glide-Schodinger dock score, which is nothing else than the EFEB (estimated free energy of binding). The docking techniques will allow us to find a specific antiviral against SARS-CoV-2, that "magic bullet" as Ehrlich said.
We would use the GlideScore programs (84)
a) The GS calculates EFEB empirically and using a heuristic approximation of the free energy by measuring ligand forces, and they have other subcomponents such as calculation of electrostatic forces, van deer Waals, and force fields. The GlideScore is used for virtual screening (85), expressed in kilocalories/mole and the more negative the value, the stronger the binding. Two subprograms called XP GlideScore and SP / HTVS GlideScore.
b) Emodel has a more significant weighting of the force field components (electrostatic and van der Waals energies), which makes it very suitable for comparing conformers, but much less suitable for comparing chemically distinct species. Therefore, Glide uses Emodel to choose the "best" pose of a ligand (pose selection) and then ranks these best positions.
c) Glide for docking uses a large class of computer algorithms that attempt to find an optimal location of a rigid or flexible ligand at the receptor binding site. Ligands are typically small molecule; peptide-protein and protein-protein docking algorithms are currently in active development. The docking algorithms also generate a score that attempts to distinguish between molecules that bind tightly at their optimal location.
d) ChemOffice+ Cloud is a robust and comprehensive suite specifically designed to simplify, facilitate and accelerate chemical communication. The cloud-native chemistry communication suite and adds access to a powerful set of tools to enable scientific research. Performs fast structure searches on all documents where chemical information is stored.
3D
e) Chimera, a 145 Mb UCSF program, free to use, not for design, but for visualization.
f) Schrödinger's Firm Programs
You use the PDB file name stored in the RCSB protein data bank and an SDF rn rl file with nearly a million lead-like compounds from a database such as ZINC in a docking program, and sit back while the computer docks each compound to each binding pocket in the protein. In reality, meaningful docking calculations require careful preparation of the receptor and ligand structures before the docking programs can do their work. With the Schrödinger suite of programs, most of the receptor preparation is done with the Protein Preparation Wizard, while ligand preparation is done with the Ligand Preparation Wizard.
g) Master Molecule Editing Program.
h) OTHERS: Autodock, EKLUSTAR, MODELLER, Chem sketch, bin RalMol, Jmol, VMD Cn-d, nanome.
CONCLUSIONS.
At present the treatment of COVID 19 is complex and multidisciplinary. It requires coordination between emergency services, intensivists and internal medicine specialists.
Drugs such as Molnupiravir or paxlovid are very interesting options, but they are not the only ones and supercomputing should continue to be used to search for specific antiviral drugs that do not generate resistance.
The global fight against this pandemic will require the efforts of both research and improvement of vaccines, as well as the development of new antivirals.(70, 71, 73, 75).
The pure Darwinian evolutionary mechanism of adaptation to the host will very possibly lead to the appearance of new escape variants, as has happened in other viruses, including DNA viruses, which mutate much less (e.g. hepatitis B), so relying solely and exclusively on mRNA vaccines seems to us to be a mistake. And we will have to have contingency plans prepared for this. We believe that screening programs for new drugs will be cost-effective and are a useful therapeutic tool to control the disease in its early stages (even subclinical) and help control cases.
We will not be able to consider the pandemic under control until the cumulative incidence is below 1 case per 100,000 inhabitants and we are far from that at the moment and this situation will last at least three more years.
The need to implement new antiviral drugs is paramount to stop the pandemic at this stage.
REFERENCES
(1)José L.Medina-Franco Jl, Fernández-de Gortaria E, Naveja J.Avances en el diseño de fármacos asistido por computadora. Progress in computer-aided drug design. Educación Química; 2015 26, (3) 180-186.
(2)Cava C, Bertoli G, Castiglioni I. In Silico Discovery of Candidate Drugs against Covid-19. Viruses. 2020 Apr 6;12(4):404. doi: 10.3390/v12040404. PMID: 32268515; PMCID: PMC7232366.
(3)Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020;252:117652. doi:10.1016/j.lfs.2020.117652
(4)Koonin, E.V., Gorbalenya, A.E. and Chumakov, K.M.(1989), Tentative identification of RNA-dependent RNA polymerases of dsRNA viruses and their relationship to positive strand RNA viral polymerases, FEBS Letters, 252, doi: 10.1016/0014-5793(89)80886-5
(5)Rabie AM. CoViTris2020 and ChloViD2020: a striking new hope in COVID-19 therapy. Mol Divers. 2021 Jan 3:1–16. doi: 10.1007/s11030-020-10169-0. Epub ahead of print. PMID: 33389560; PMCID: PMC7778709.
(6) Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020;252:117652. doi:10.1016/j.lfs.2020.117652
(7)Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020;252:117652. doi:10.1016/j.lfs.2020.117652
(8)Alnajjar R, Mostafa A, Kandeil A, Al-Karmalawy AA. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon. 2020 Dec;6(12):e05641. doi: 10.1016/j.heliyon.2020.e05641. Epub 2020 Dec 3. PMID: 33294721; PMCID: PMC7713577.
(9)Wu J , Wang H , Li B . Structure-aided ACEI-capped remdesivir-loaded novel PLGA nanoparticles: toward a computational simulation design for anti-SARS-CoV-2 therapy. Phys Chem Chem Phys. 2020 Dec 23;22(48):28434-28439. doi: 10.1039/d0cp04389c. PMID: 33305304.
(10)Keretsu, S., Bhujbal, S.P. & Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci Rep 10, 17716 (2020). https://doi.org/10.1038/s41598-020-74468-
(11)Zhenming Jin, Xiaoyu Du, Yechun Xu, Yongqiang Deng, Meiqin Liu, Yao Zhao, Bing Zhang, Xiaofeng Li, Leike Zhang, Yinkai Duan, Jing Yu, Lin Wang, Kailin Yang, Fengjiang Liu, Tian You, Xiaoce Liu, Xiuna Yang, Fang Bai, Hong Liu, Xiang Liu, Luke W. Guddat, Gengfu Xiao, Chengfeng Qin, Zhengli Shi, Hualiang Jiang, Zihe Rao, Haitao YangbioRxiv 2020.02.26.964882; doi: https://doi.org/10.1101/2020.02.26.964882
(12) Mohapatra S, Nath P, Chatterjee M, Das N, Kalita D, et al. (2020) Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLOS ONE 15(11): e0241543. https://doi.org/10.1371/journal.pone.0241543
(13)Mohapatra S, Nath P, Chatterjee M, Das N, Kalita D, et al. (2020) Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLOS ONE 15(11): e0241543. https://doi.org/10.1371/journal.pone.0241543
(14)El Hassab Mahmoud A., Shoun Aly A., Al-Rashood T., Al-Warhi Tarfah, Identification of a New Potential SARS-COV-2 RNA-Dependent RNA Polymerase Inhibitor via Combining Fragment-Based Drug Design, Docking, Molecular Dynamics, and MM-PBSA Calculations JFrontiers in Chemistry.2020; 8:915-916. DOI=10.3389/fchem.2020.584894
(15)K. Abraham Peele, Chandrasai Potla Durthi, T. Srihansa, S. Krupanidhi, Vijaya Sai Ayyagari, D. John Babu, M. Indira, A. Ranganadha Reddy, T.C. Venkateswarulu,Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study,Informatics in Medicine Unlocked; 2020,(19)100-345ISSN 2352-9148,https://doi.org/10.1016/j.imu.2020.100345.
(16) Giwa AL, Desai A, Duca A. Novel 2019 coronavirus SARS-CoV-2 (COVID-19): An updated overview for emergency clinicians. Emerg Med Pract. 2020 May 1;22(5):1-28. Epub 2020 Mar 24. PMID: 32207910. ( figura de las 4 proteinas9
(17) Peele KA, Potla Durthi C, Srihansa T, Krupanidhi S, Ayyagari VS, Babu DJ, Indira M, Reddy AR, Venkateswarulu TC. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform Med Unlocked. 2020;19:100345. doi: 10.1016/j.imu.2020.100345. Epub 2020 May 11. PMID: 32395606; PMCID: PMC7211761.
(18) Li F. Structure, function, and evolution of coronavirus spike proteins. Annual Rev. Virol. 2016; 3(1):237‐261.
(19)Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. China Life Sci. 2020; 63(3):457‐460.
(20) Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. 2020;579(7798):270‐273.
(21)Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Science Alliance. 2020;3(9):e202000786.
(22)Hoffmann M, Hofmann‐Winkler H, Smith JC, et al. Camostat mesylate inhibits SARS‐CoV‐2 activation by TMPRSS2‐related proteases and its metabolite GBPA exerts antiviral activity. bioRxiv 2020.
(23)Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik O, Rohde C, Klenk HD, Garten W, Steinmetzer T, Böttcher-Friebertshäuser E. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. 2020 Jul 23;3(9):e202000786. doi: 10.26508/lsa.202000786. PMID: 32703818; PMCID: PMC7383062.
(24)Walls AC, Tortorici MA, Bosch B‐J, et al. Cryo‐electron microscopy structure of a coronavirus spike glycoprotein trimer. 2016; 531(7592): 114‐ 117.
(25) Gomes CP, Fernandes DE, Casimiro F, da Mata GF, Passos MT, Varela P, Mastroianni-Kirsztajn G, Pesquero JB. Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front Cell Infect Microbiol. 2020 Dec 8;10:589505. doi: 10.3389/fcimb.2020.589505. PMID: 33364201; PMCID: PMC7753008.
(26)Breining P, Frølund AL, Højen JF, Gunst JD, Staerke NB, Saedder E, Cases-Thomas M, Little P, Nielsen LP, Søgaard OS, Kjolby M. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin Pharmacol Toxicol. 2021 Feb;128(2):204-212. doi: 10.1111/bcpt.13533. Epub 2020 Nov 22. PMID: 33176395.
(27)Hydroxychloroquine and Azithromycin as a treatment of COVID-19: preliminary results of an open-label non-randomized clinical trial medRxiv (2020) doi: 10.1101/2020.03.16.20037135International Journal of Antimicrobial Agents (2020) doi: 10.1016/j.ijantimicag.2020.105949
(28)Heidi Ledford, Richard Van Noorden, «High-profile coronavirus retractions raise concerns about data oversight,» Nature (05 Jun 2020), doi: https://doi.org/10.1038/d41586-020-01695-w
(29)Prinz JHWu HSarich Met al.Markov models of molecular kinetics: generation and validation.J Chem Phys. 2011; 134174105
(30) Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein Alexandra C. Walls, Young-Jun Park, M. Alexandra Tortorici, Abigail Wall, Andrew T. McGuire, David VeeslerbioRxiv 2020.02.19.956581; doi: https://doi.org/10.1101/2020.02.19.956581
(31)Cryo-EM structure of S-Trimer, a subunit vaccine candidate for COVID-19Jiahao Ma, Danmei Su, Xueqin Huang, Ying Liang, Yan Ma, Peng Liang, Sanduo ZhengbioRxiv 2020.09.21.306357; Rxiv preprint 306357 (21 Sep 2020) https://doi.org/10.1101/2020.09.21.306357
(32)Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, Liu X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J Med Chem. 2020 Nov 12; 63(21):12256-12274. doi: 10.1021/acs.jmedchem.0c00502. Epub 2020 Jun 25. PMID: 32539378; PMCID: PMC7315836.
(33)Lan, J., Ge, J., Yu, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature (2020). https://doi.org/10.1038/s41586-020-2180-5).
(34)Shang, J., Ye, G., Shi, K. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020). https://doi.org/10.1038/s41586-020-2179-y
(35) Lan, J., Ge, J., Yu, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020). https://doi.org/10.1038/s41586-020-2180-5
(36)Shang, J., Ye, G., Shi, K. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020). https://doi.org/10.1038/s41586-020-2179-y
(37) Wenfei Song, Miao Gui, Ye Xiang, «Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2, » PLoS Pathogens, 14: e1007236 (13 Aug 2018), doi: https://doi.org/10.1371/journal.ppat.1007236.
(38)Ludovico Cantuti-Castelvetri, Ravi Ojha, Liliana D. Pedro, Minou Djannatian, Jonas Franz, Suvi Kuivanen, Katri Kallio, Tuğberk Kaya, Maria Anastasina, Teemu Smura, Lev Levanov, Leonora Szirovicza, Allan Tobi, Hannimari Kallio-Kokko, Pamela Österlund, Merja Joensuu, Frédéric A. Meunier, Sarah Butcher, Martin Sebastian Winkler, Brit Mollenhauer, Ari Helenius, (39)Ozgun Gokce, Tambet Teesalu, Jussi Hepojoki, Olli Vapalahti, Christine Stadelmann, Giuseppe Balistreri, Mikael Simons NRP1 Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system bioRxiv 2020.06.07.137802; doi: https://doi.org/10.1101/2020.06.07.137802
(40) Neuropilin-1 is a host factor for SARS-CoV-2 infectionJames L. Daly, Boris Simonetti, Carlos Antón-Plágaro, Maia Kavanagh Williamson, Deborah K. Shoemark, Lorena Simón-Gracia, Katja Klein, Michael Bauer, Reka Hollandi, Urs F. Greber, Peter Horvath, Richard B. Sessions, Ari Helenius, Julian A. Hiscox, Tambet Teesalu, David A. Matthews, Andrew D. Davidson, Peter J. Cullen, Yohei YamauchibioRxiv 2020.06.05.134114; doi: https://doi.org/10.1101/2020.06.05.134114
(41)Padmanabhan, Pranesh; Desikan, Rajat; Dixit, Narendra (2020): Targeting TMPRSS2 and Cathepsin B/L Together May Be Synergistic Against SARS-CoV-2 Infection. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.12213125.v2
(42)Ruoslahti «RGD and other recognition sequences for integrins» Annu. Rev. Cell Dev. Biol. 1996, 12: 697–715».
(43)Friess H, Kleeff J, Isenmann R, Malfertheiner P, Büchler MW. Adaptation of the human pancreas to inhibition of luminal proteolytic activity. Gastroenterology. 1998; 115(2): 388‐ 396.
(44)Ohkoshi M, Oka T. Clinical experience with a protease inhibitor [N, N‐dimethylcarbamoylmethyl 4‐ (4‐guanidinobenzoyloxy) ‐phenylacetate] methanesulfate for prevention of recurrence of carcinoma of the mouth and in treatment of terminal carcinoma. J. Maxillofacial Surg. 1984; 12(4): 148‐ 152.
(45)Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015; 116: 76‐ 84.
(46)Shirato K, Kawase M, Matsuyama S. Middle east respiratory syndrome coronavirus infection mediated by the trans membrane serine protease TMPRSS2. J Virol. 2013; 87(23): 12552‐ 12561.
(47)Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012; 86(12): 6537‐ 6545.
(48)Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. Protease‐mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci. 2005; 102(35): 12543‐ 12547.
(49)Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012; 86(12): 6537‐ 6545.
(50)Reagan‐Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659‐661.
(51)Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP‐mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875‐894.
(52)Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76‐84.
(53)Midgley I, Hood AJ, Proctor P, et al. Metabolic fate of 14C‐camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica. 1994; 24(1): 79‐ 92.
(54) Ohki SNishiyama H Ozeki KIto HHirata F.Studies on absorption, distribution, metabolism and excretion of [14C] FOY-305.Gendai-Iryo. 1980; 12: 71-82
(55)Senokuchi K, Nakai H, Nakayama Yet al. New orally active serine protease inhibitors.J Med Chem. 1995; 38: 2521-2523
(56) Beckh KGoke BMuller RArnold R.Elimination of the low-molecular weight proteinase inhibitor camostate (FOY 305) and its degradation products by the rat liver.Res Exp Med (Berl). 1987; 187: 401-406
(57) Beckh KWeidenbach HWeidenbach FMuller RAdler G.Hepatic and pancreatic metabolism and biliary excretion of the protease inhibitor camostat mesilate.Int J Pancreatol. 1991; 10: 197-205
(58) Senokuchi KNakai HNakayama Yet al.New orally active serine protease inhibitors.J Med Chem. 1995; 38: 2521-2523
(59)Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020; 181(2): 271‐ 280.e8.
(60)Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Science Alliance. 2020; 3(9):e202000786.
(61)Yamaya M, Shimotai Y, Hatachi Y, et al. The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm Pharmacol Ther. 2015; 33: 66‐ 74.
(62)Hempel T Raich L Olsson Set al. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat.Chemical Science. 2021;
(63)Midgley I, Hood AJ, Proctor P, et al. Metabolic fate of 14C‐camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica. 1994; 24(1): 79‐ 92.
(64)Sun W, Zhang X, Cummings MD, et al. Targeting enteropeptidase with reversible covalent inhibitors to achieve metabolic benefits. Journal of Pharmacology and Experimental Therapeutics. 2020; JPET– AR. http://dx.doi.org/10.1124/jpet.120.000219.
(65) Spraggon G, Hornsby M, Shipway A, et al. Active site conformational changes of prostasin provides a new mechanism of protease regulation by divalent cations. Protein Sci. 2009; 18(5): 1081‐ 1094.
(66) Hempel T, Raich L, Olsson S, et al. Molecular mechanism of SARS‐CoV‐2 cell entry inhibition via TMPRSS2 by Camostat and Nafamostat mesylate. bioRxiv 2020:2020.07.21.214098 https://doi.org/10.1101/2020.07.21.214098
(67) Depfenhart M, de Villiers D, Lemperle G, Meyer M, Di Somma S. Potential new treatment strategies for COVID‐19: is there a role for bromhexine as add‐on therapy? Intern Emerg Med. 2020; 15(5): 801‐ 812.
(68)13/20 Informationen der Institutionen und Behörden: BMG: Zentrale Beschaffung vonArzneimitteln zur Therapie schwerwiegender Verläufe COVID-19 infizierter Patienten undVerteilung an Apotheken durch die Bundeswehr (24.03.2020). 2020 [cited 7/9/2020]; Availablefrom: https://www.abda.de/fuer-apotheker/arzneimittelkommission/amk-nachrichten/detail/13-20-informationen-der-institutionen-und-behoerden-bmg-zentrale-beschaffung-von-arzneimittelnzur-therapie-schwerwiegender-verlaeufe-covid-19-infizierter-patienten-und-verteilung-anapotheken-durch-die-bundeswehr/
(69)Hofmann-Winkler H, Moerer O, Alt-Epping S, Bräuer A, Büttner B, Müller M, et al. CamostatMesylate but Not Hydroxychloroquine May Reduce Severity of Organ Failure in COVID-19Sepsis: A Case-Control Study (6/15/2020) 2020
(70)Ji W, Huh K, Kang M, Hong J, Bae GH, Lee R, et al. Effect of Underlying Comorbidities on theInfection and Severity of COVID-19 in Korea: a Nationwide Case-Control Study. J Korean MedSci. 2020 Jun 29;35(25):e237.
(71)Yamamoto M, Kiso M, Sakai-Tagawa Y, Iwatsuki-Horimoto K, Imai M, Takeda M, Kinoshita N, Ohmagari N, Gohda J, Semba K, Matsuda Z, Kawaguchi Y, Kawaoka Y, Inoue JI. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses. 2020 Jun 10;12(6):629. doi: 10.3390/v12060629. PMID: 32532094; PMCID: PMC7354595.
(72)Ou T, Mou H, Zhang L, Ojha A, Choe H, Farzan M (2021) Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog 17(1): e1009212. doi:10.1371/journal.ppat.1009212
(73)Maurizio Guastalegname, Alfredo Vallone, Could Chloroquine /Hydroxychloroquine Be Harmful in Coronavirus Disease 2019 (COVID-19) Treatment?, Clinical Infectious Diseases, Volume 71, Issue 15, 1 August 2020, Pages 888–889, https://doi.org/10.1093/cid/ciaa321
(74)Siyu Xiu, Alexej Dick, Han Ju, Sako Mirzaie, Fatemeh Abdi, Simon Cocklin, Peng Zhan, and Xinyong Liu Inhibitors of SARS-CoV-2 Entry: Current and Future OpportunitiesJournal of Medicinal Chemistry 2020 63 (21), 12256-12274DOI: 10.1021/acs.jmedchem.0c00502
https://www.bbc.com/mundo/noticias-internacional-55406204
(75)Song W, Gui M, Wang X, Xiang Y (2018) Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 14(8): e1007236. https://doi.org/10.1371/journal.ppat.1007236
(76)Hoffmann et al., Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and itsmetabolite GBPA exerts antiviral activity, EBioMedicine (2021), https://doi.org/10.1016/j.ebiom.2021.103255
(77)Barnard DL, Day CW, Bailey K, et al. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir Chem Chemother 2006; 17:275–84.
https://www.clinicaltrials.gov/ct2/show/NCT04381936
https://www.clinicaltrialsregister.eu/
(https://www.clinicaltrials.gov/ct2/show/NCT04381936
https://www.clinicaltrials.gov/ct2/show/NCT04321616
https://trialsitenews.com/study-combines-korean-pancreatitisdrug-foistar-with-
https://www.medrxiv.org/content/10.1101/2






